Pushing the boundaries of population genomic computation with the tree sequence
Center for Genome Research and Biocomputing
Oregon State // 26 May 2021
Inverse problems

Inverse problems
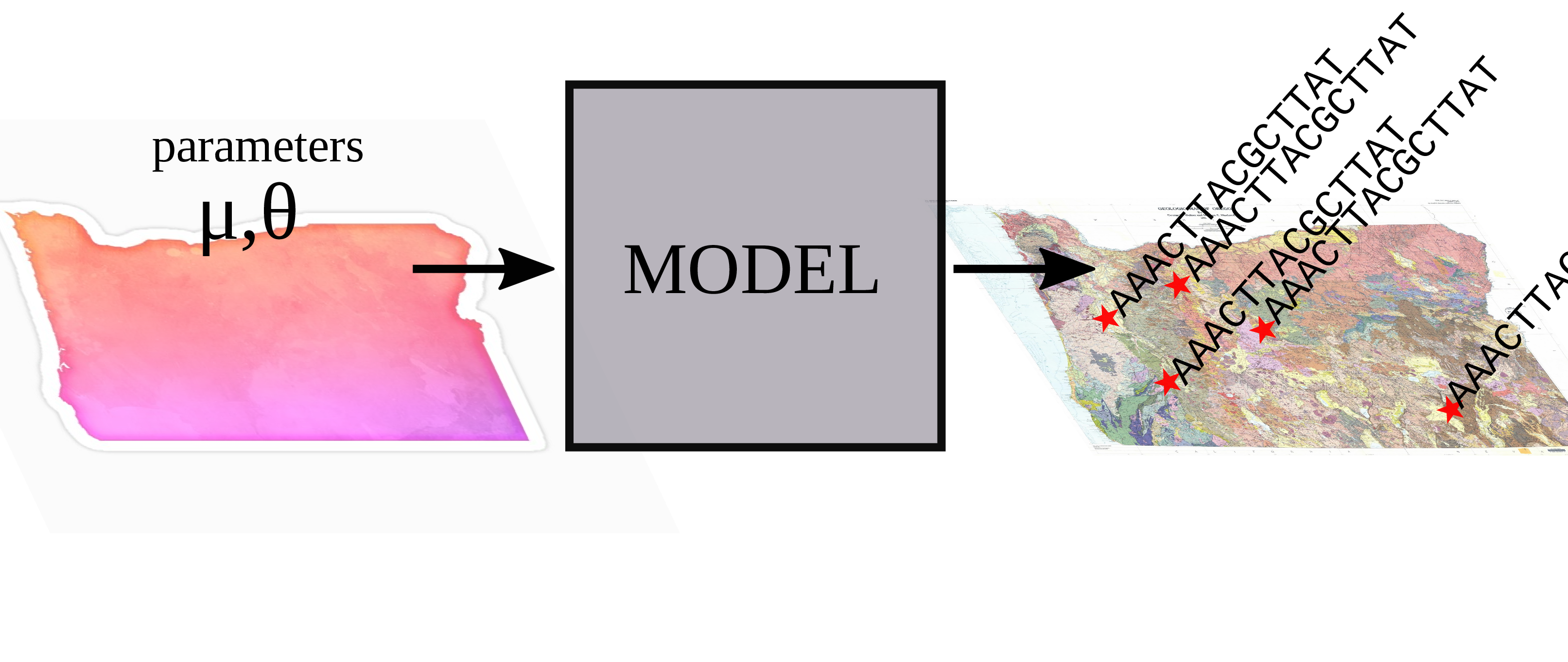
Inverse problems
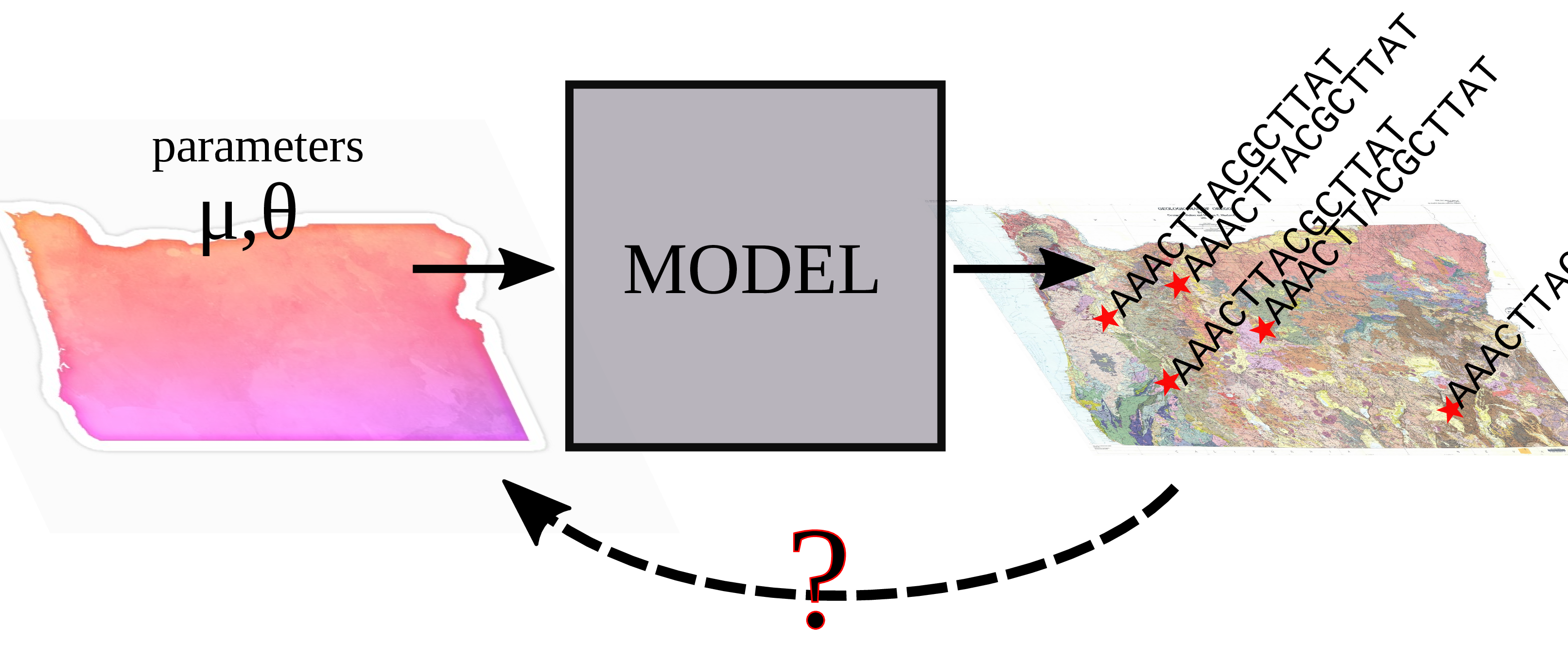
Inverse problems
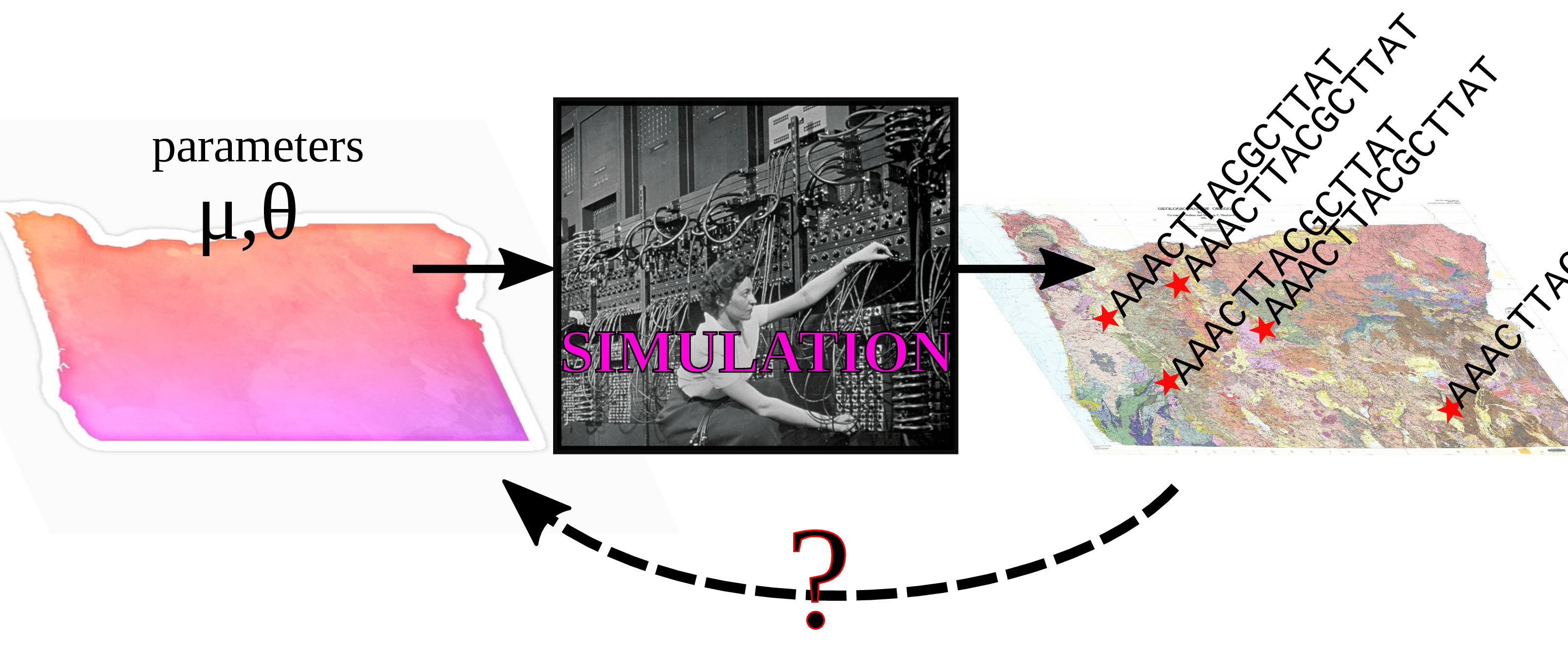
Simulation-based inference
- bespoke confirmatory simulations
- optimization of one or two parameters
- machine learning predictors (e.g., random forests)
- Approximate Bayesian Computation (ABC)
- deep learning
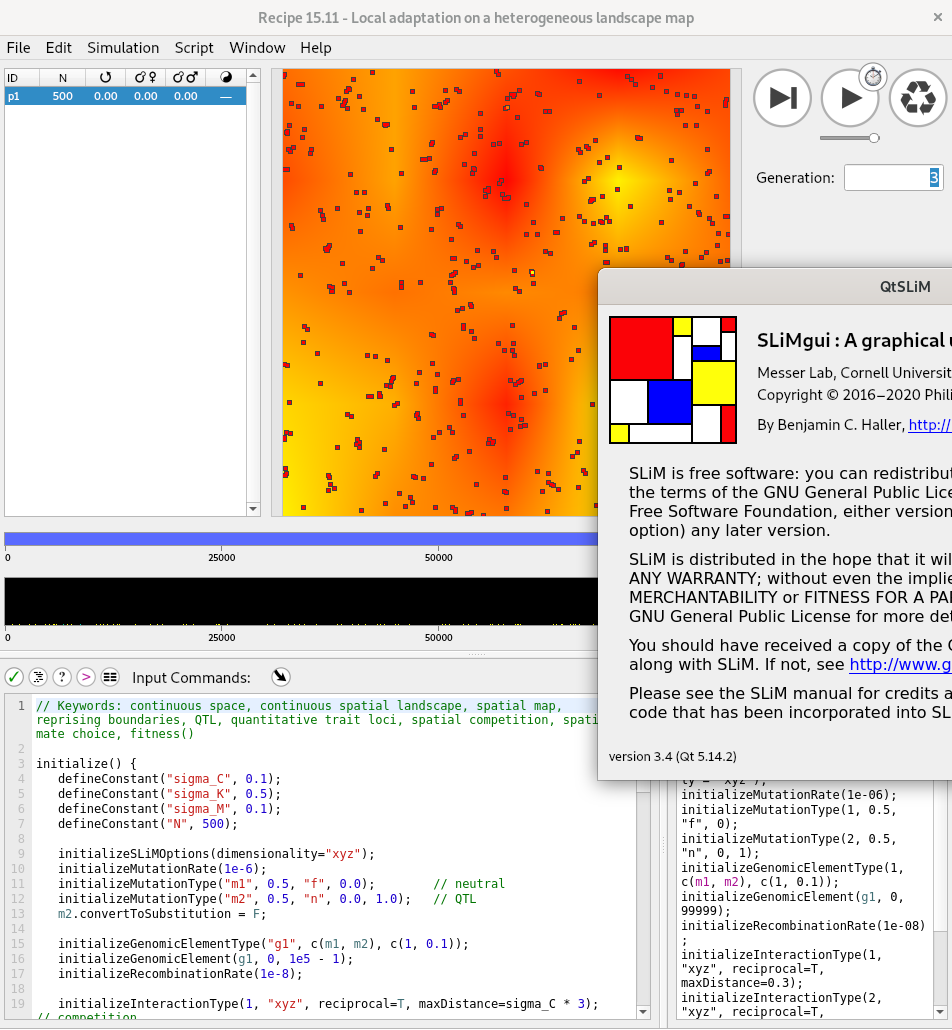
Enter SLiM
by Ben Haller and Philipp Messer
- a forwards simulator
- arbitary life cycles
- continuous geography and local interactions
- quantitative traits
- anything is possible
 Ben Haller
Ben Haller
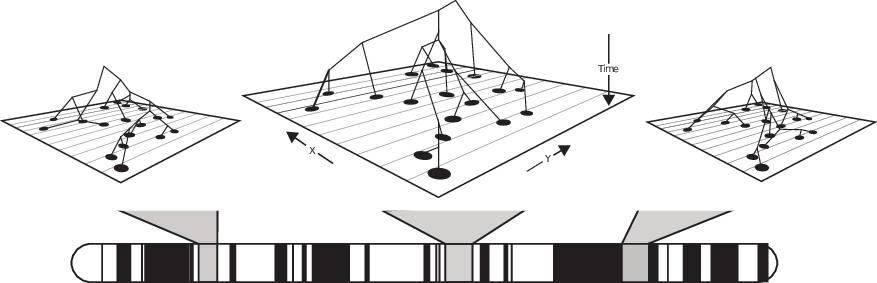
History is a sequence of trees
For a set of sampled chromosomes, at each position along the genome there is a genealogical tree that says how they are related.

The succinct tree sequence
is a way to succinctly describe this, er, sequence of trees
and the resulting genome sequences.

jerome kelleher
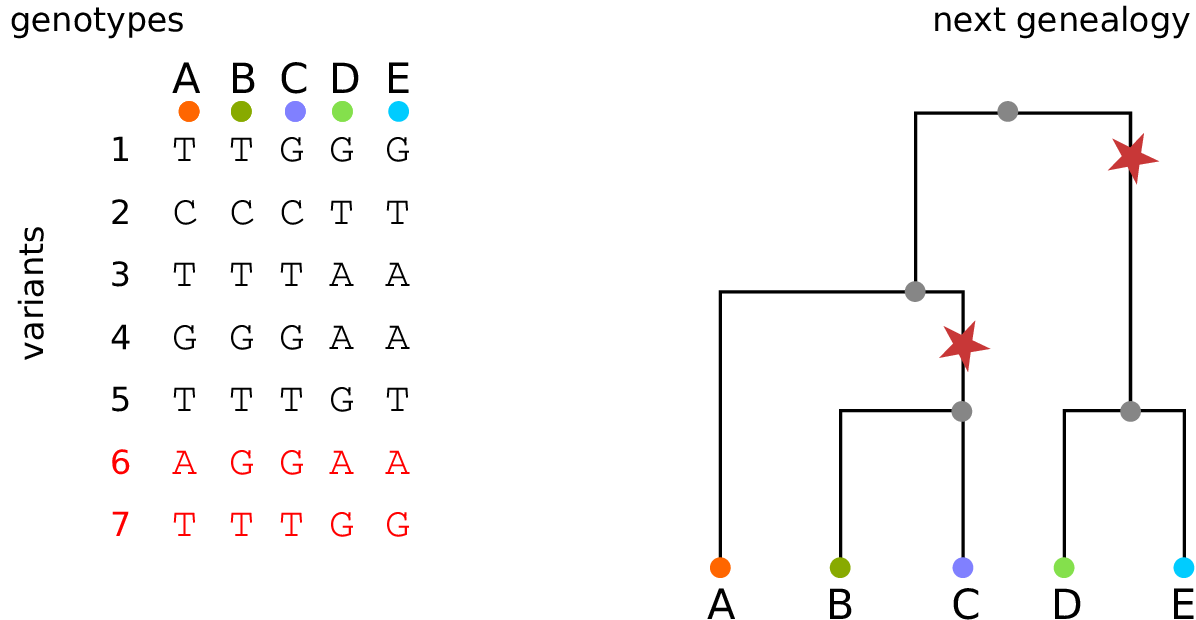
Example: three samples; two trees; two variant sites
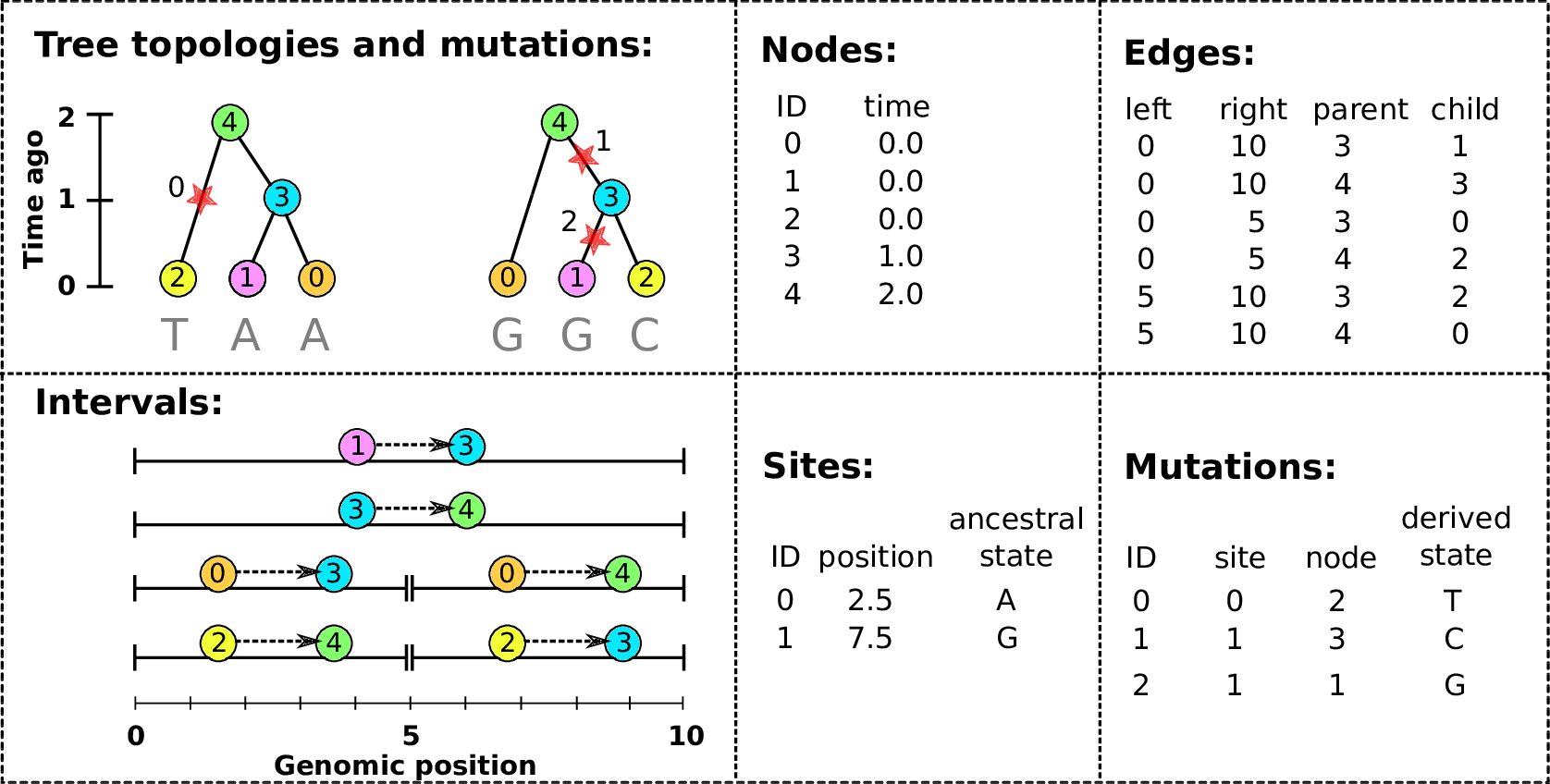
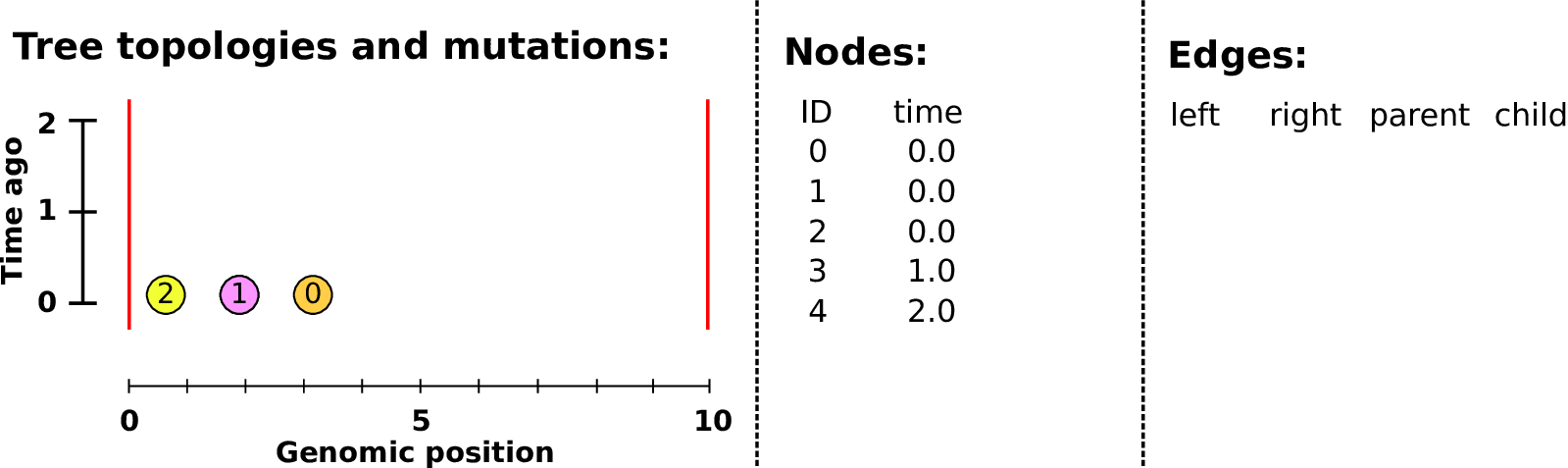
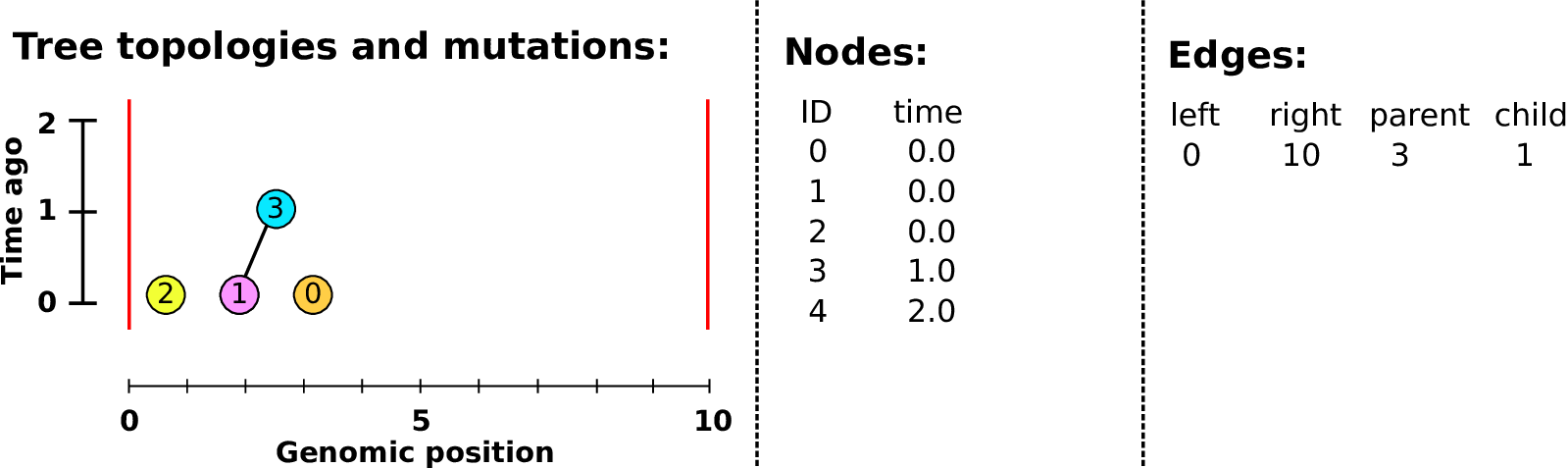
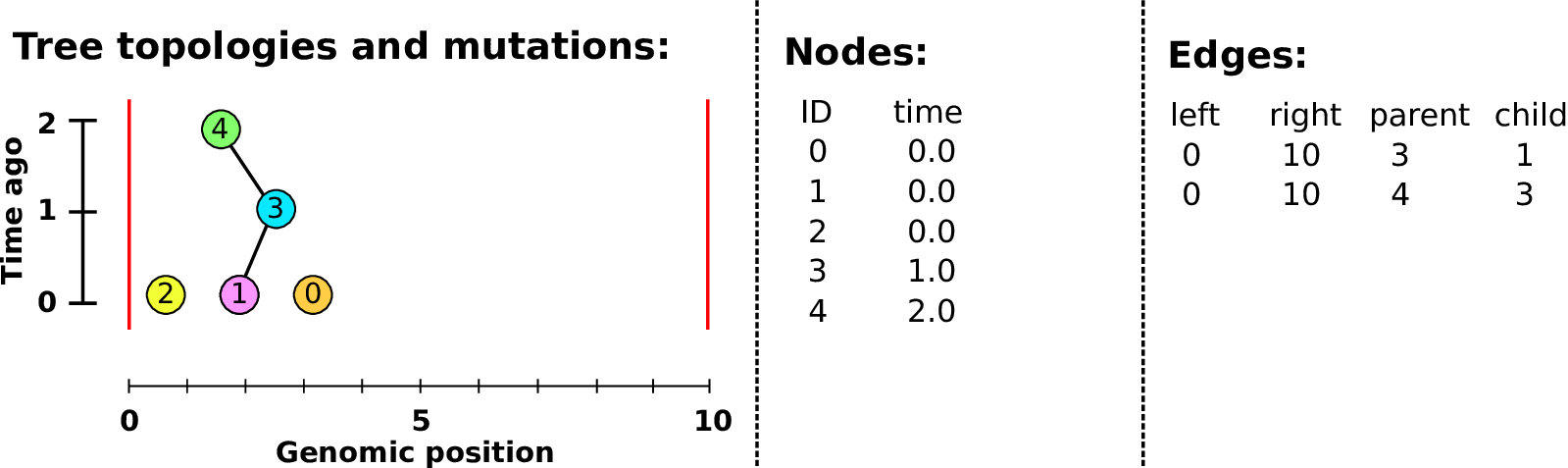
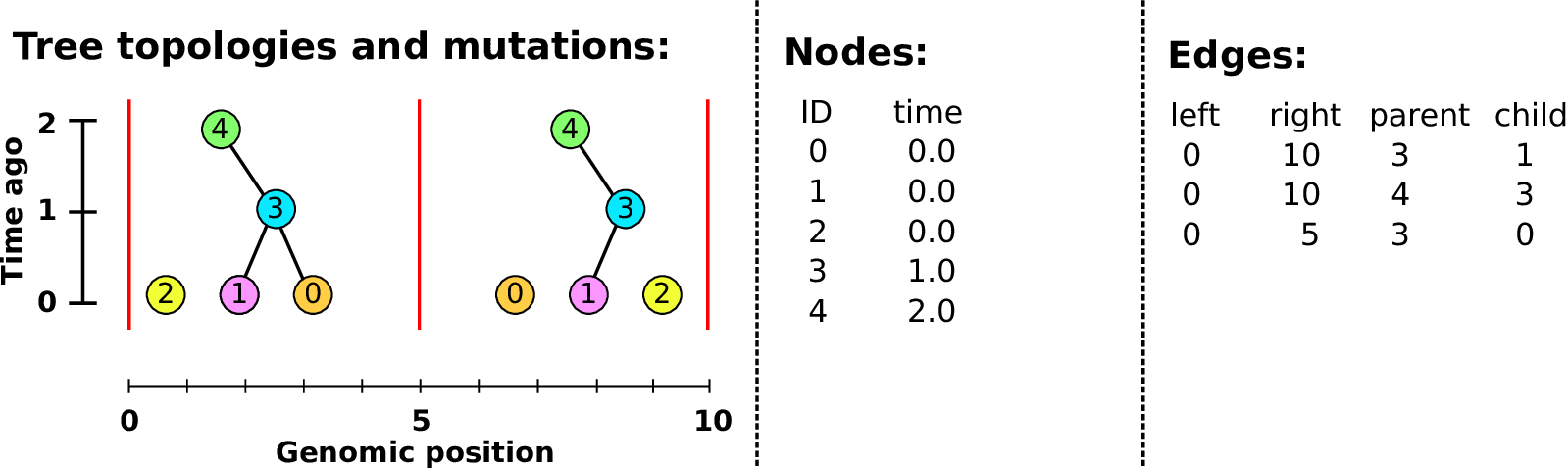
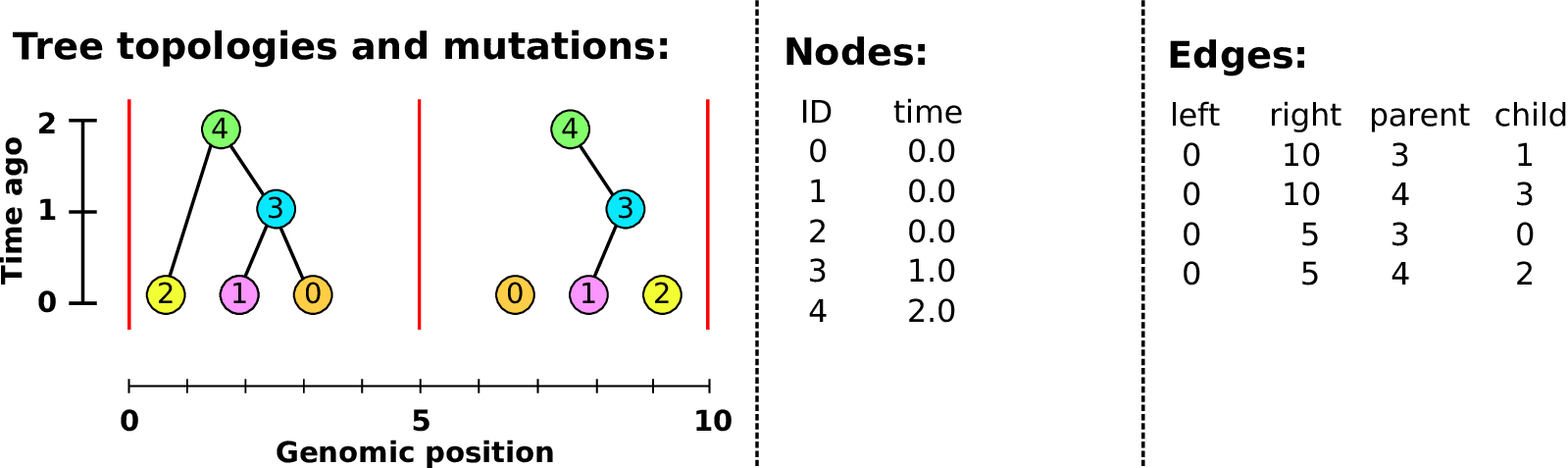
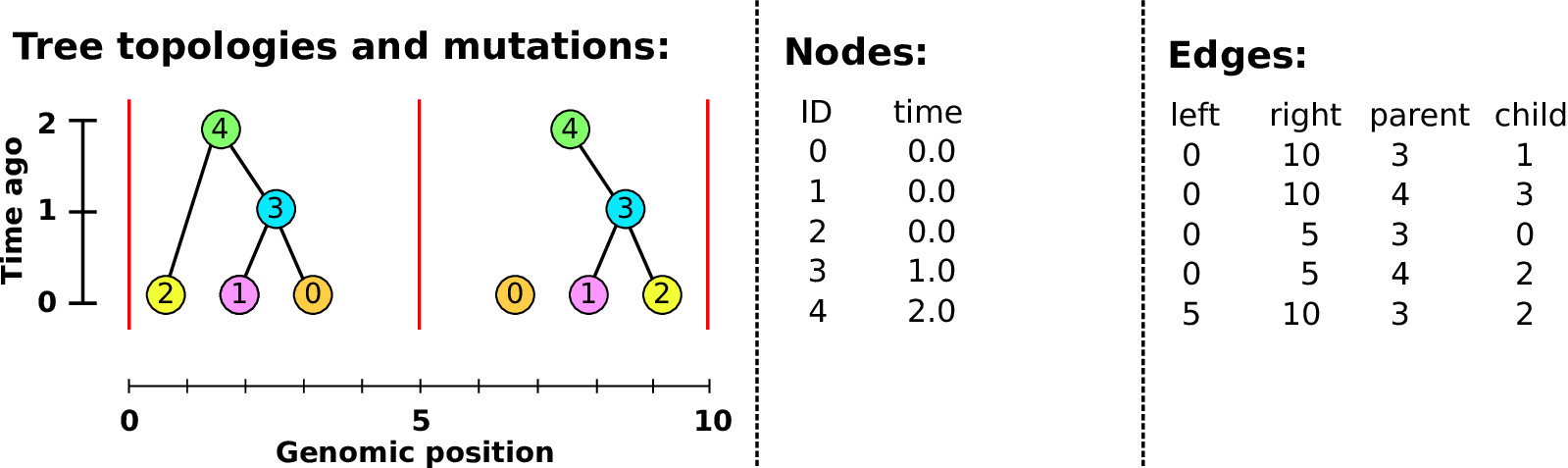
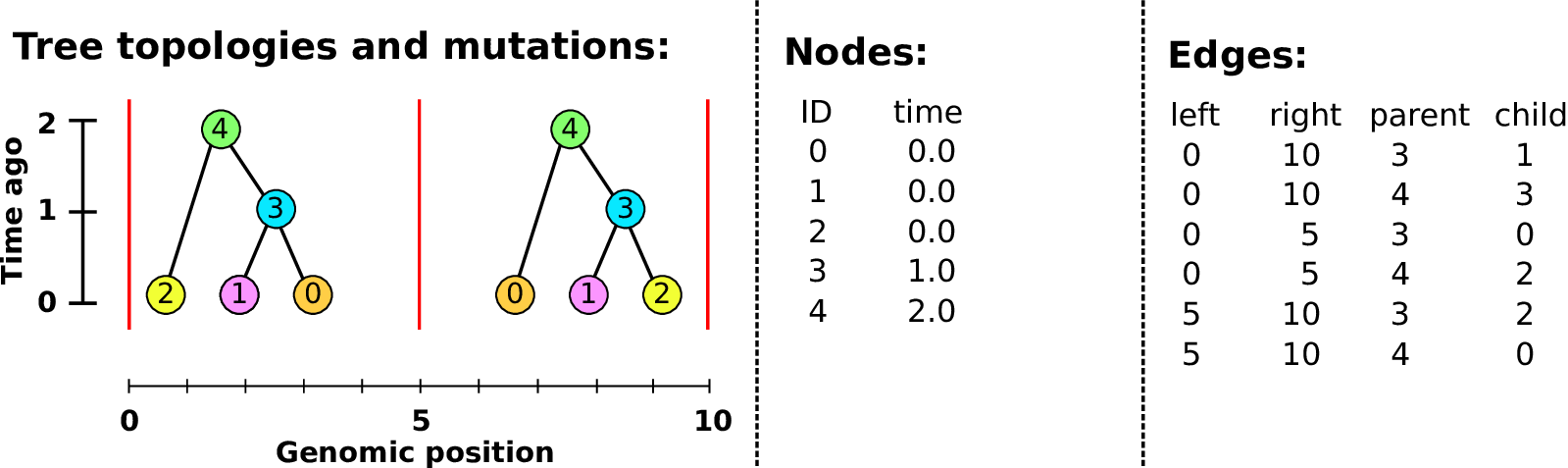
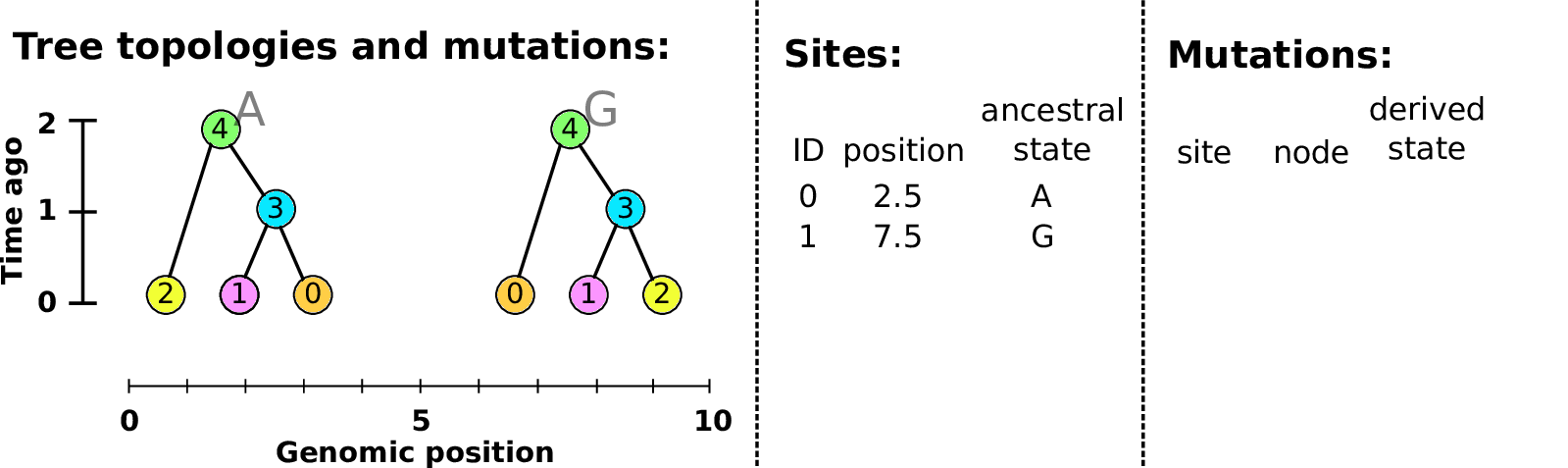
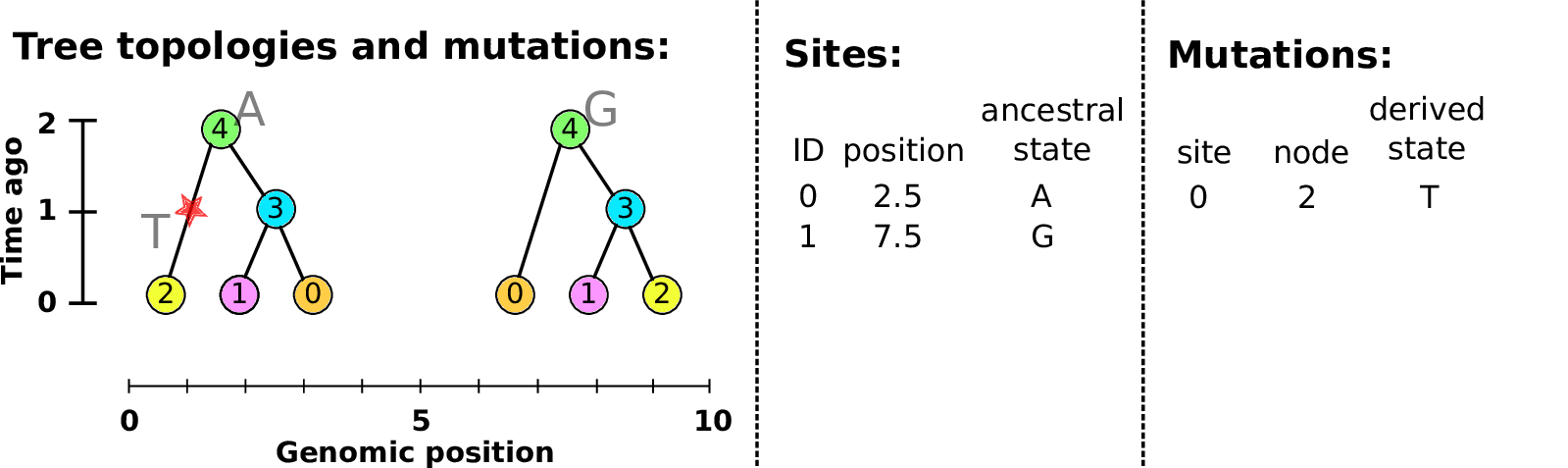
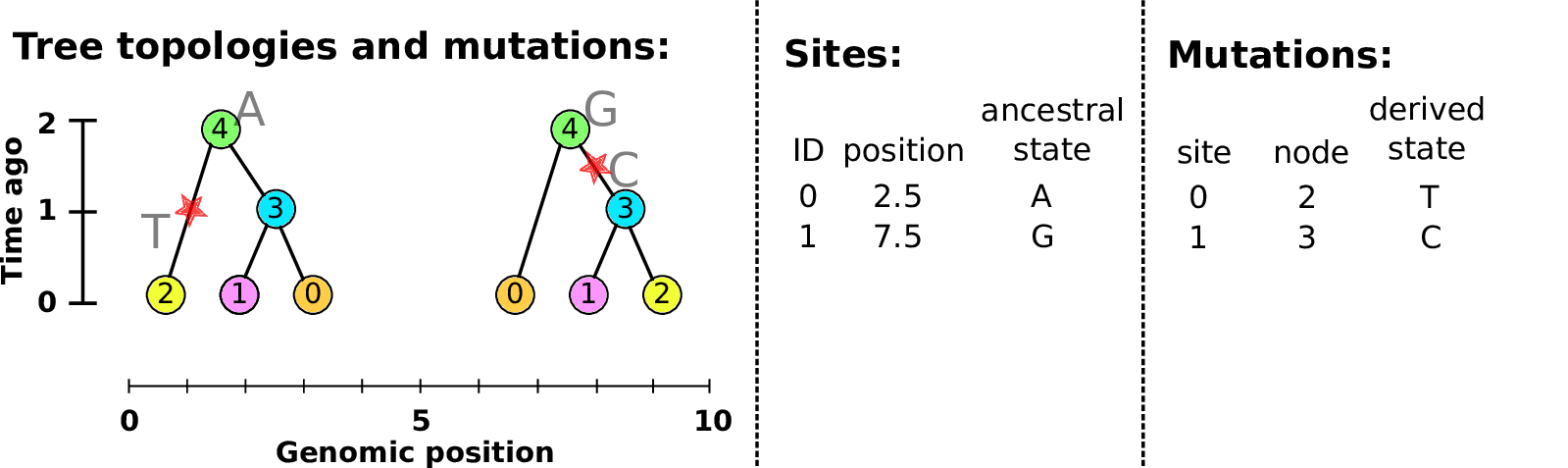
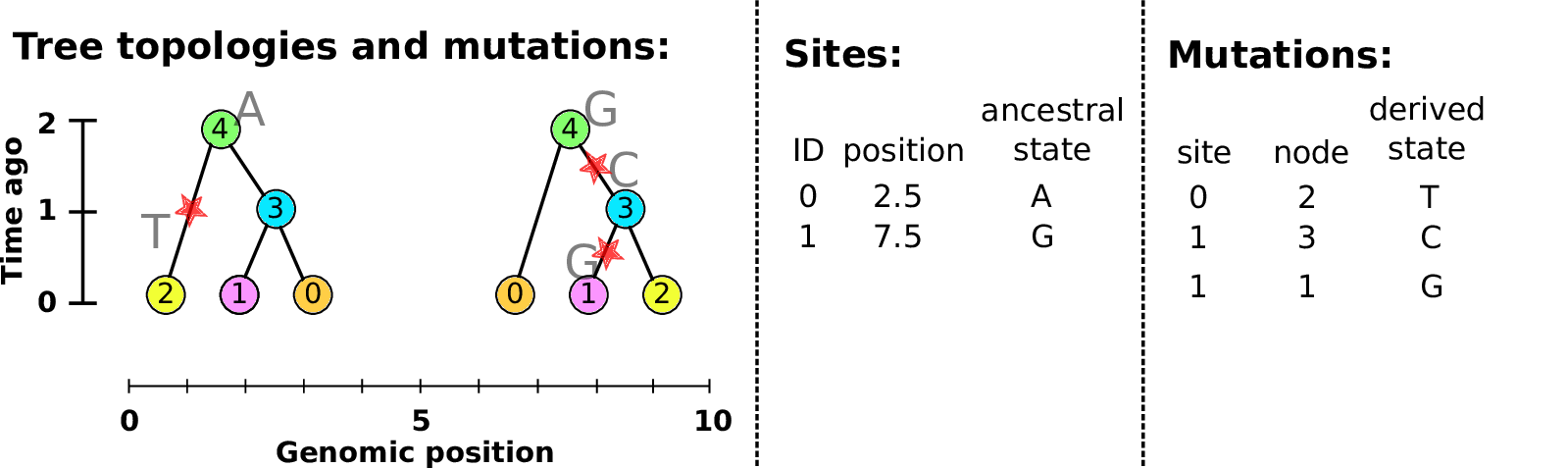
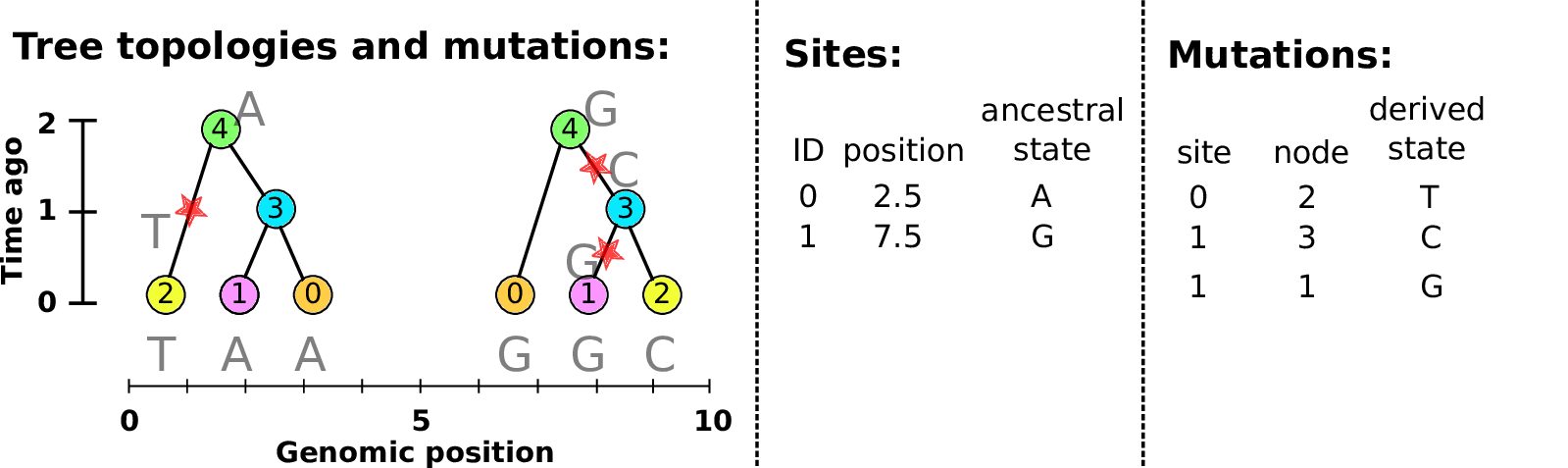
The result: an encoding of the genomes and all the genealogical trees.
File sizes
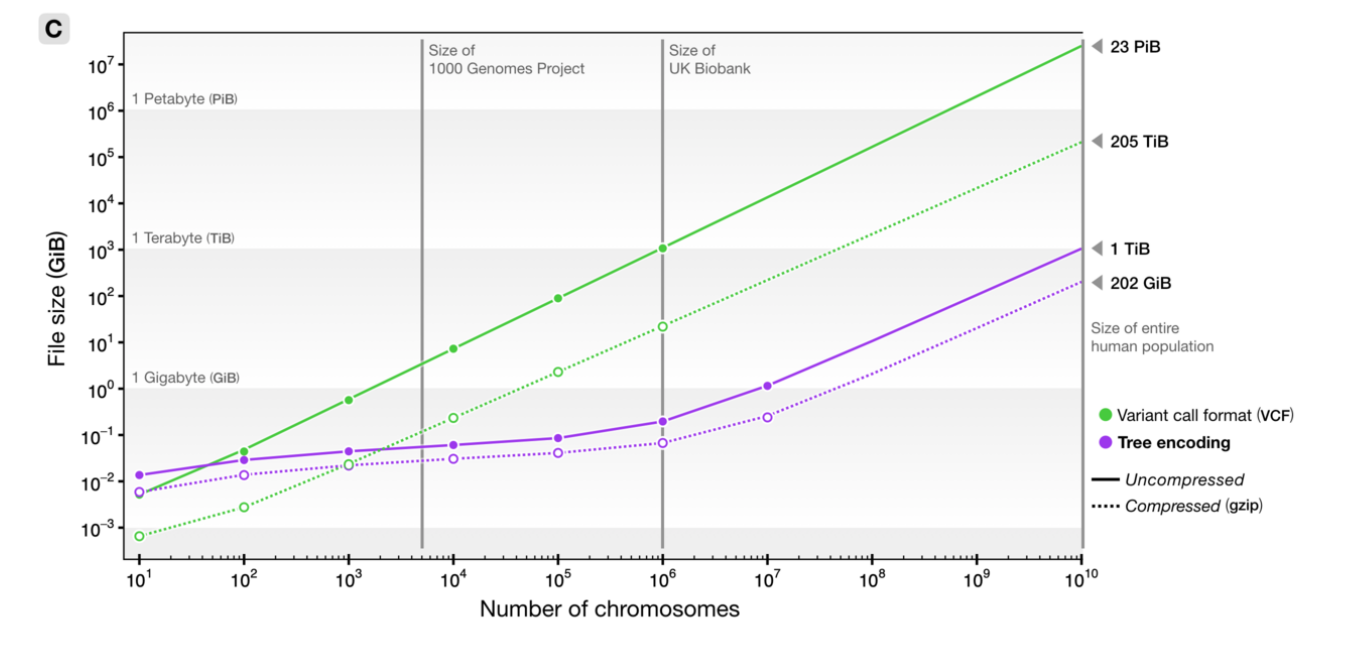
100Mb chromosomes; from Kelleher et al 2018, Inferring whole-genome histories in large population datasets, Nature Genetics
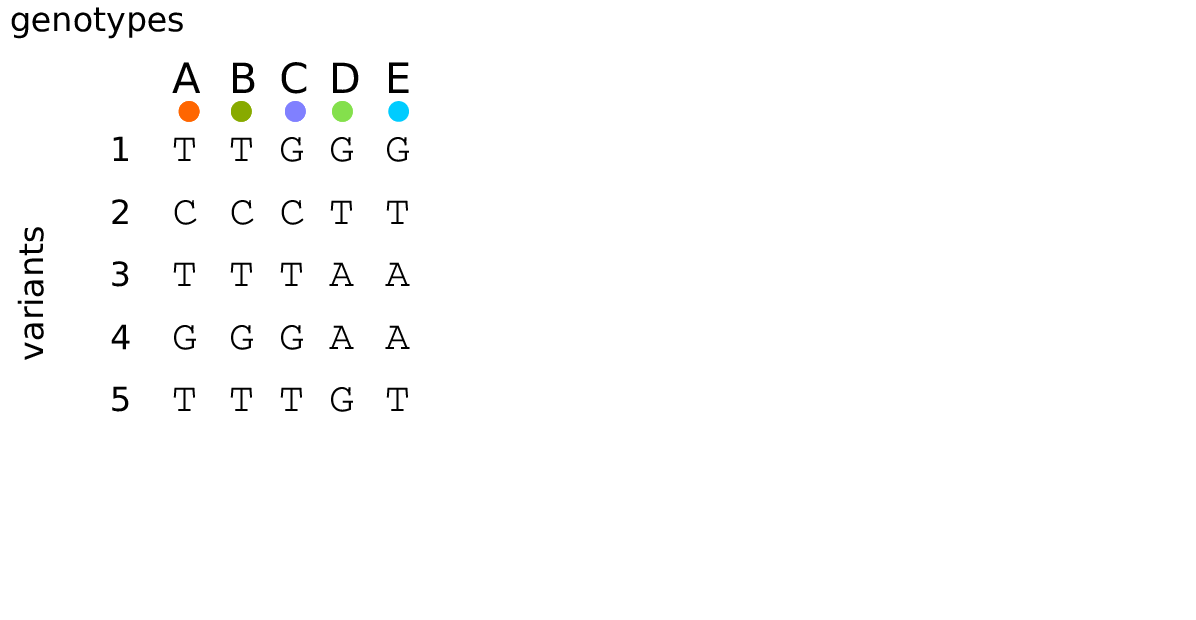
For \(N\) samples genotyped at \(M\) sites
Genotype matrix:
\(N \times M\) things.
Tree sequence:
- \(2N-2\) edges for the first tree
- \(\sim 4\) edges per each of \(T\) trees
- \(M\) mutations
\(O(N + T + M)\) things
Fast genotype statistics, too!
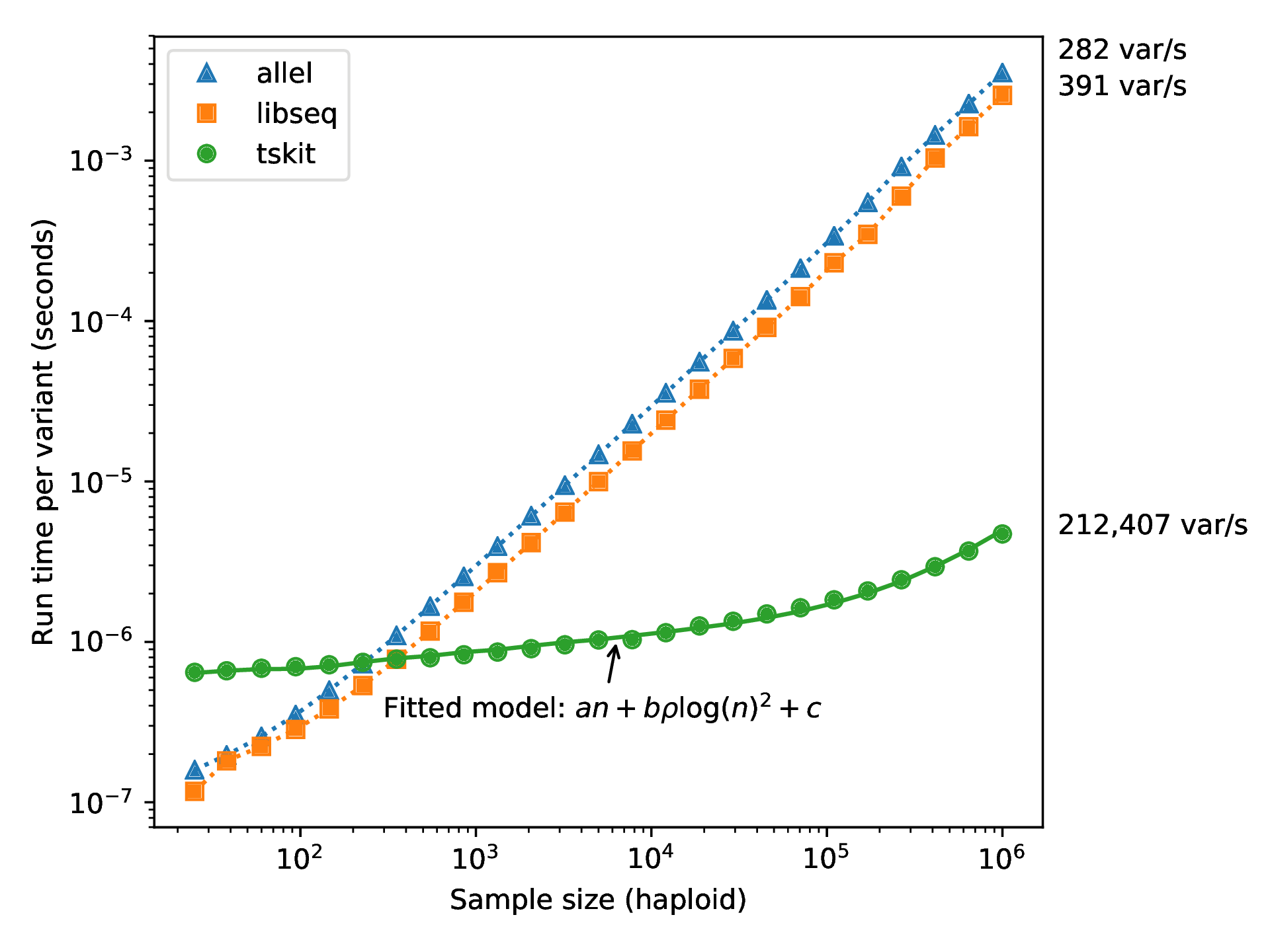
from Ralph, Thornton and Kelleher 2019, Efficiently summarizing relationships in large samples
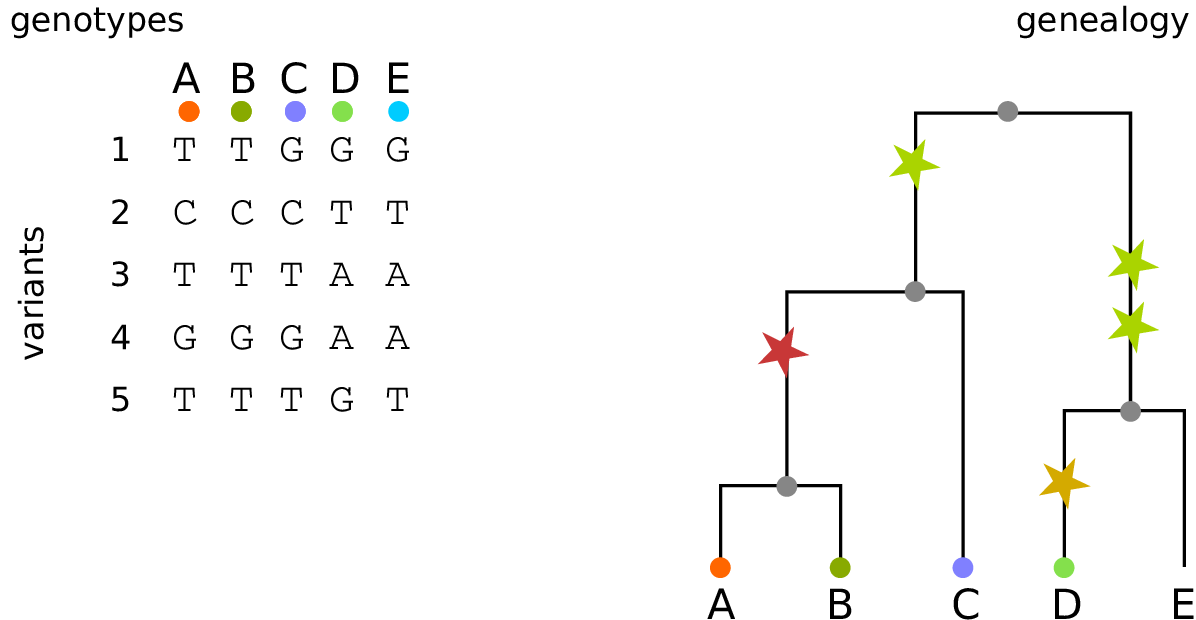
The main idea
If we record the tree sequence that relates everyone to everyone else,
after the simulation is over we can put neutral mutations down on the trees.
Since neutral mutations don’t affect demography,
this is equivalent to having kept track of them throughout.
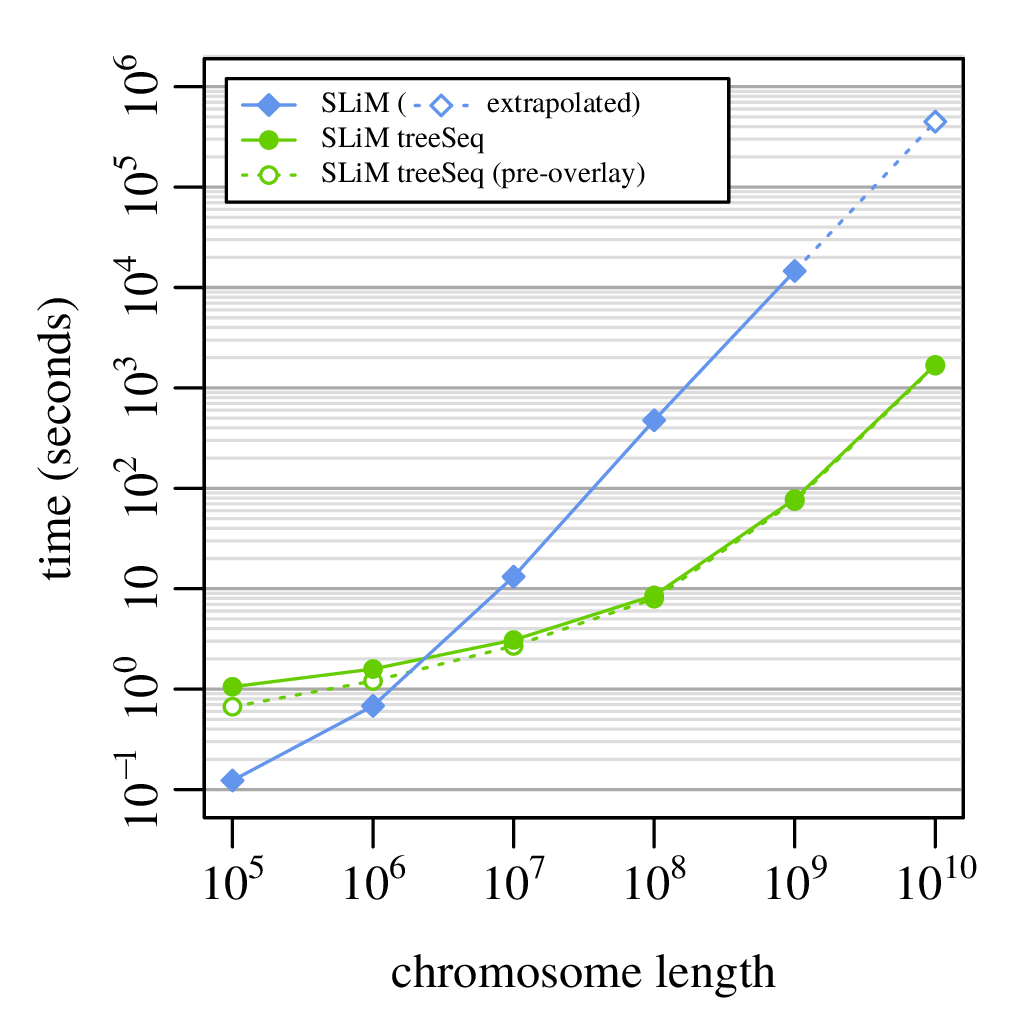
From Kelleher, Thornton, Ashander, and Ralph 2018, Efficient pedigree recording for fast population genetics simulation.
and Haller, Galloway, Kelleher, Messer, and Ralph 2018, Tree‐sequence recording in SLiM opens new horizons for forward‐time simulation of whole genomes


A 100x speedup!
Example 1: landscapes of diversity
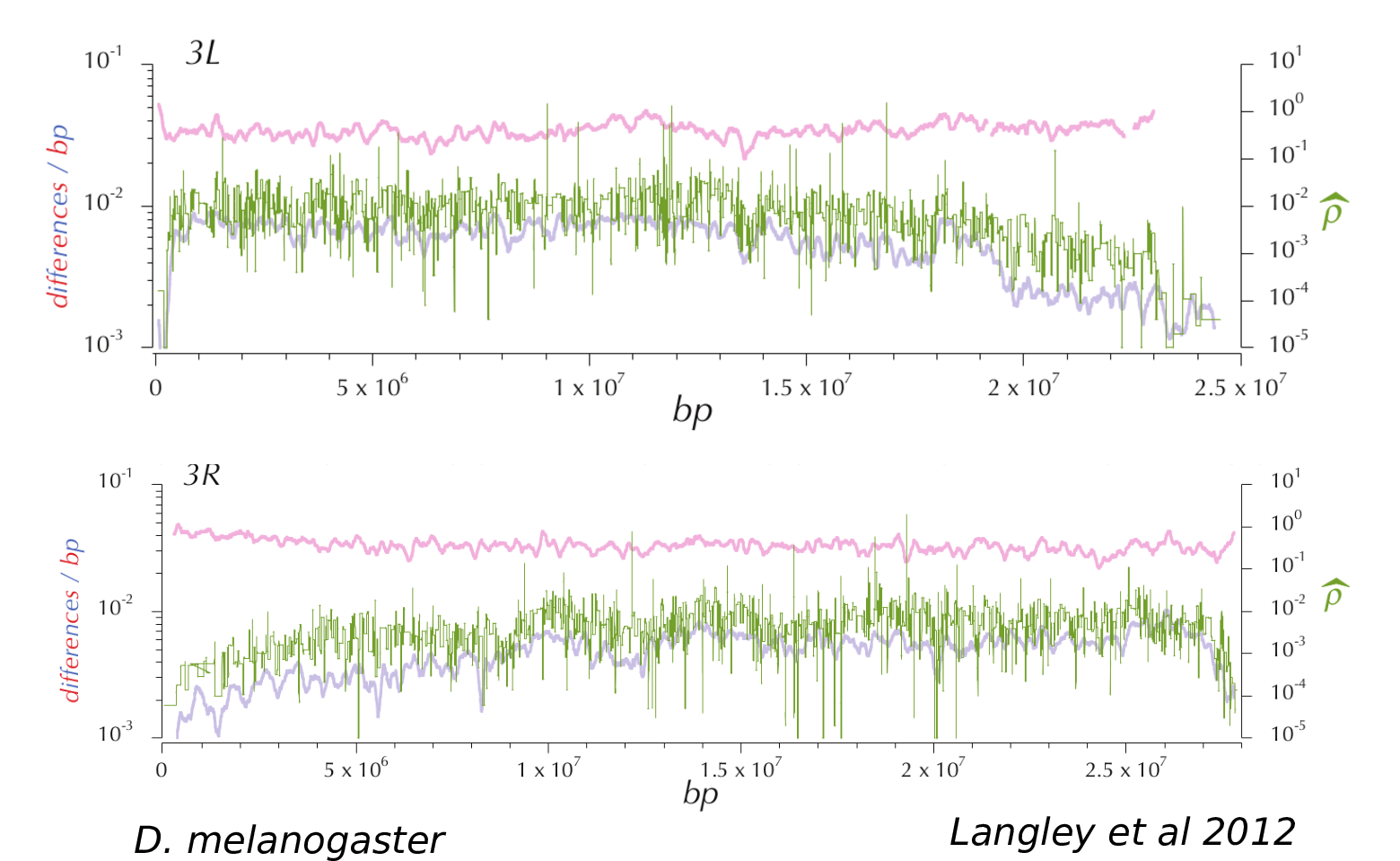
Diversity correlates with recombination rate
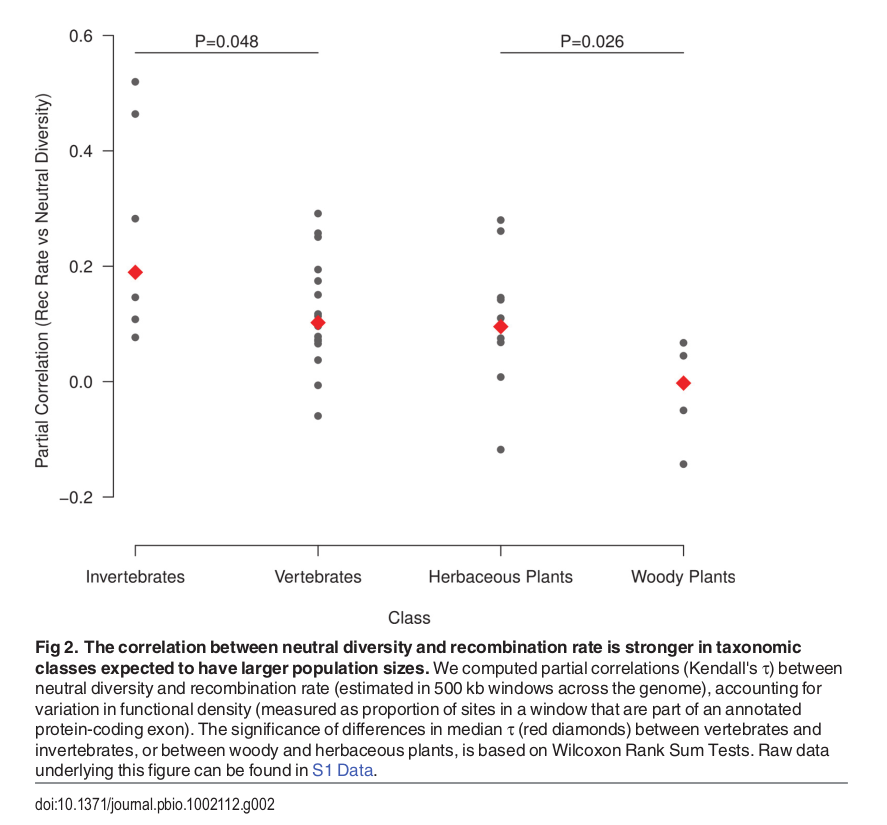
Hudson 1994; Cutter & Payseur 2013; Corbett-Detig et al 2015
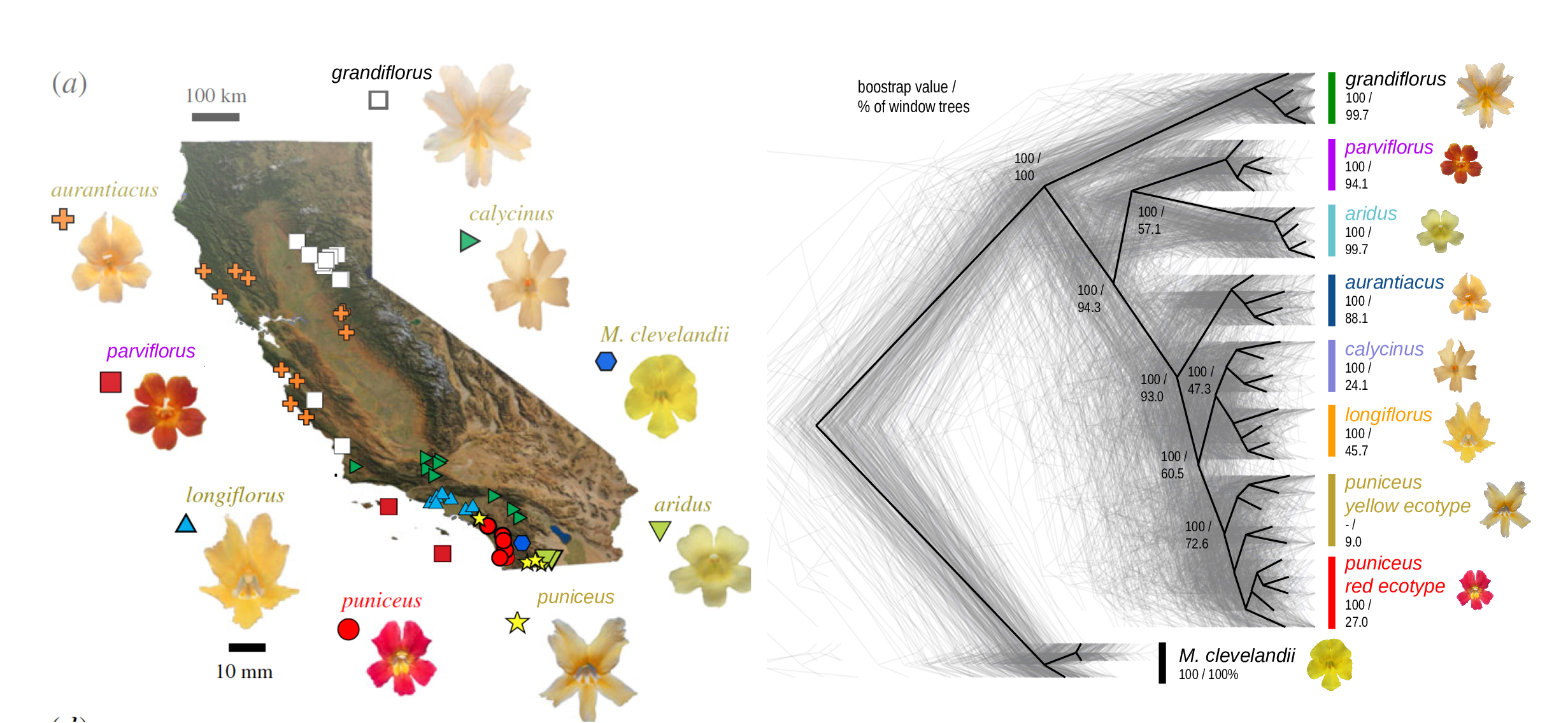
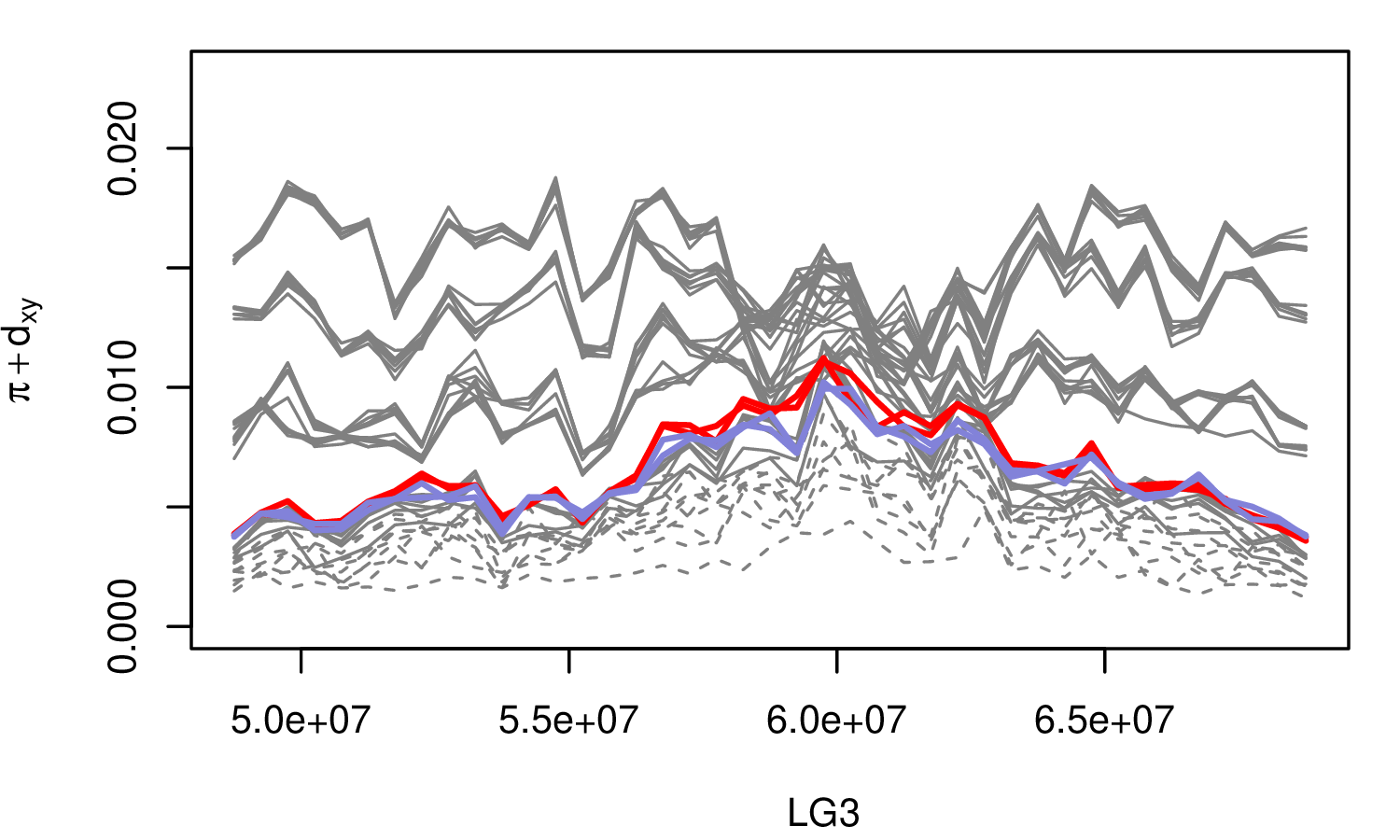
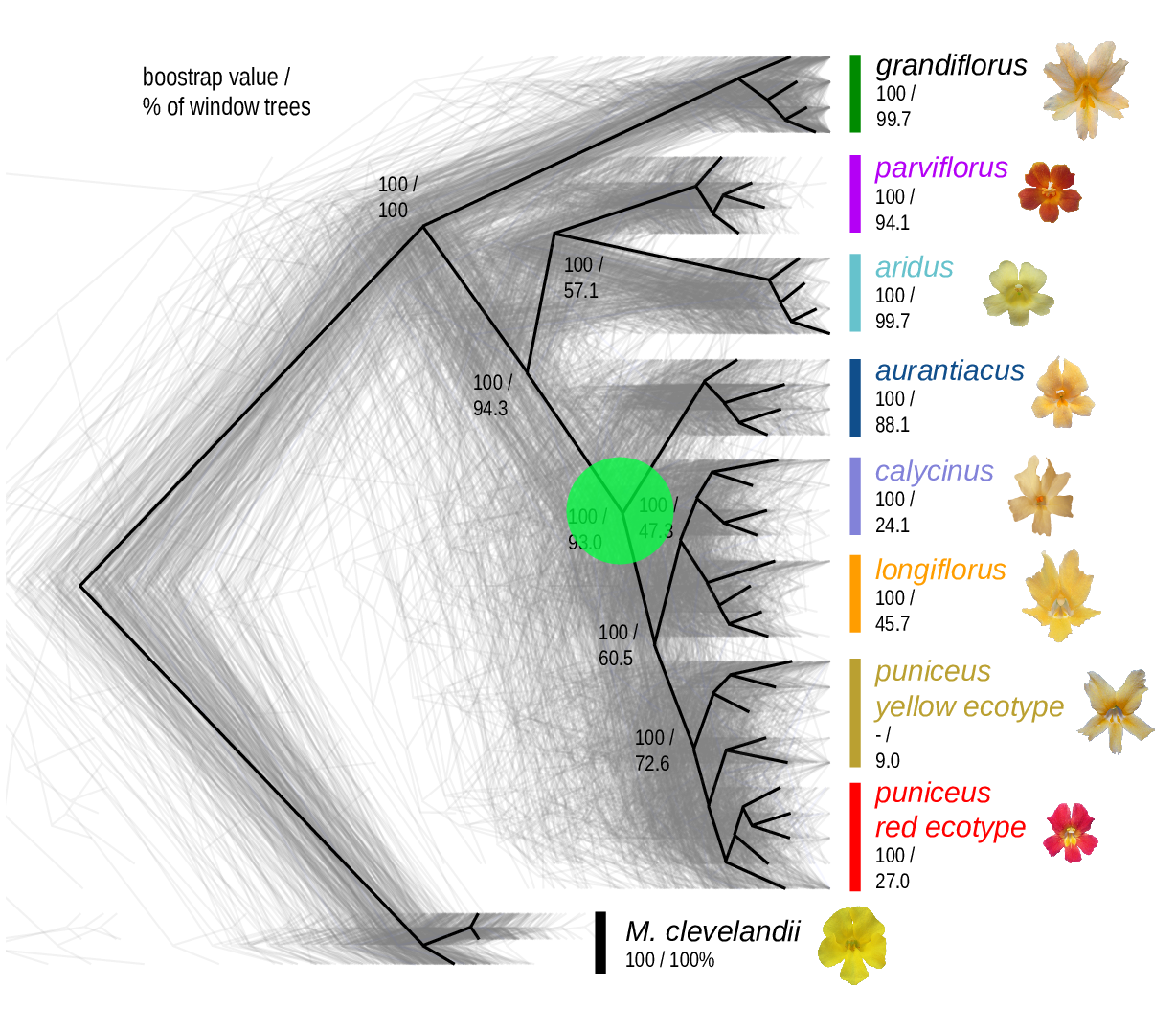
The Mimulus aurantiacus species complex
Simulations
\(N=10,000\) diploids
burn-in for \(10N\) generations
population split, with either:
- neutral
- background selection
- selection against introgressed alleles
- positive selection
- local adaptation

Murillo Rodrigues

From Widespread selection and gene flow shape the genomic landscape during a radiation of monkeyflowers, Stankowski, Chase, Fuiten, Rodrigues, Ralph, and Streisfeld; PLoS Bio 2019.
Example 2: identifying sweeps

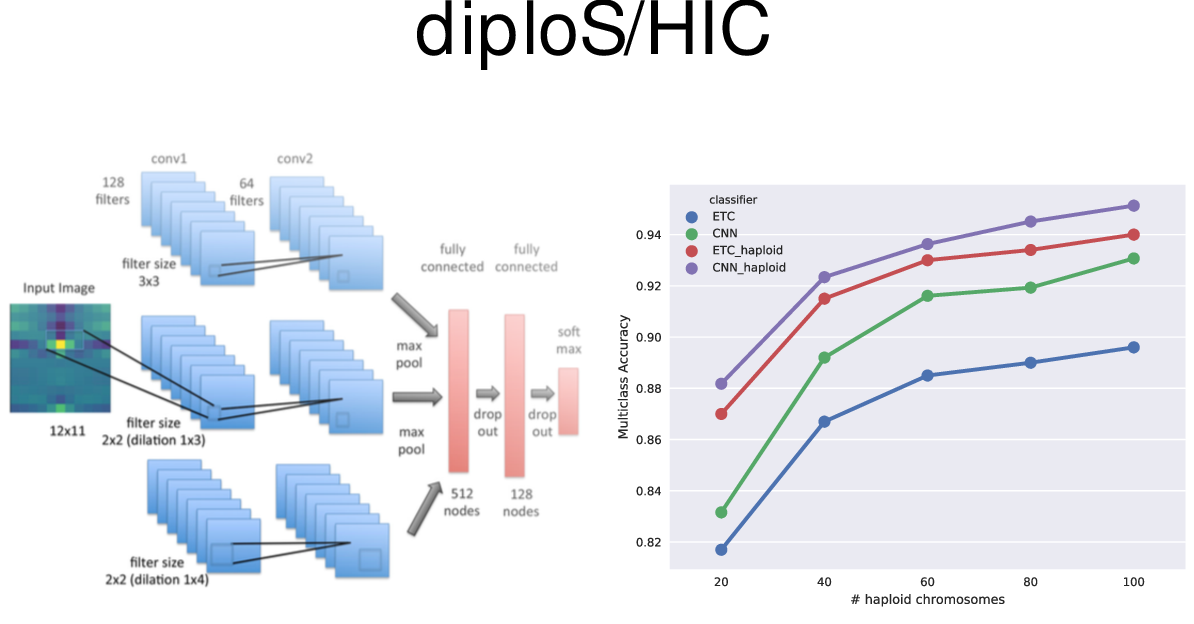
Example 3: predicting location
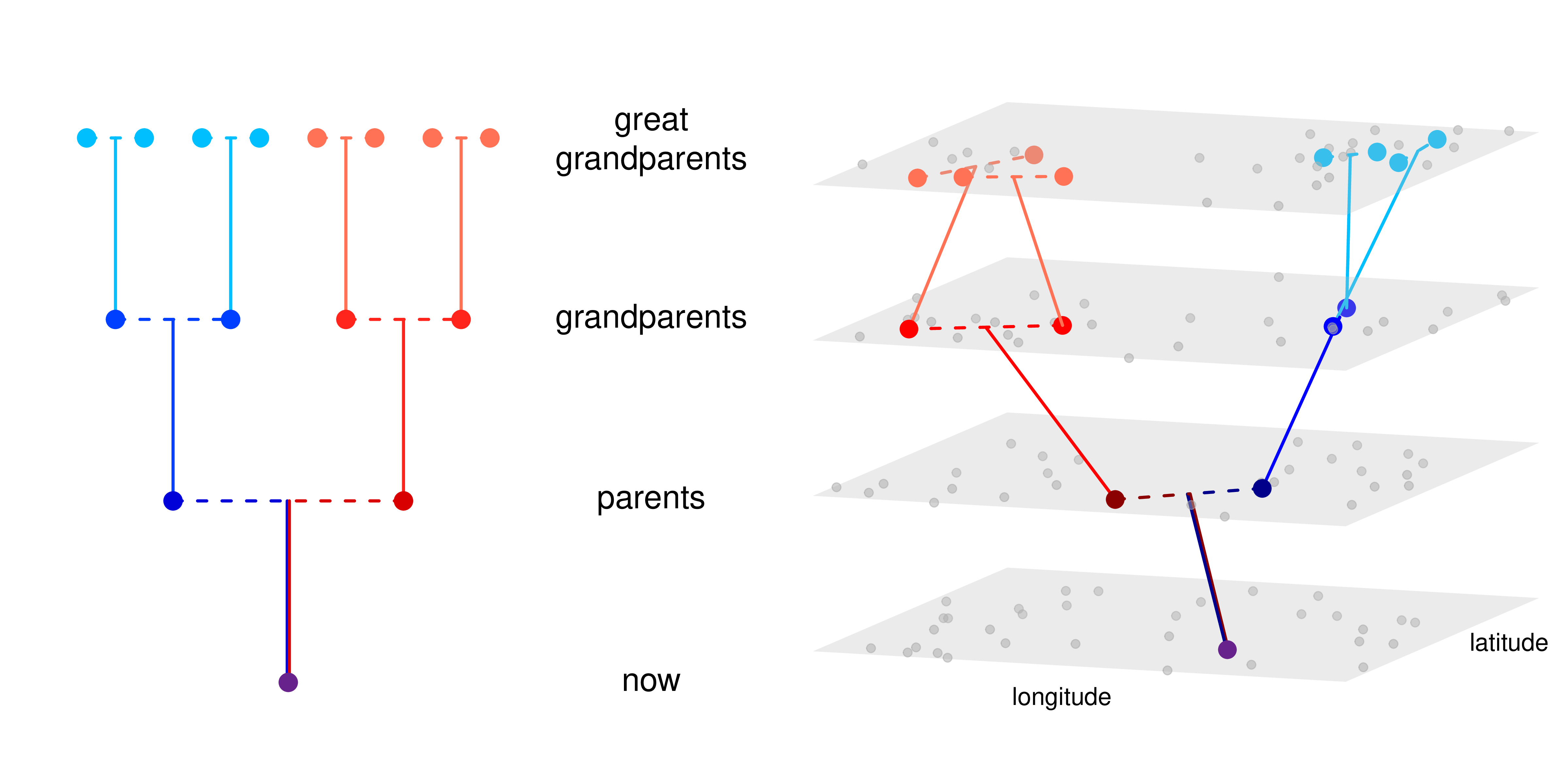
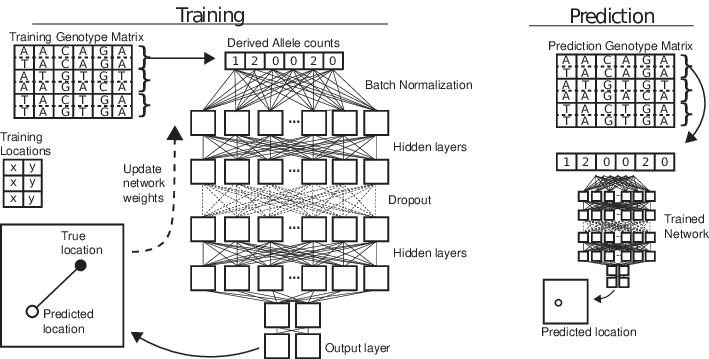

locator (Battey et al 2020)
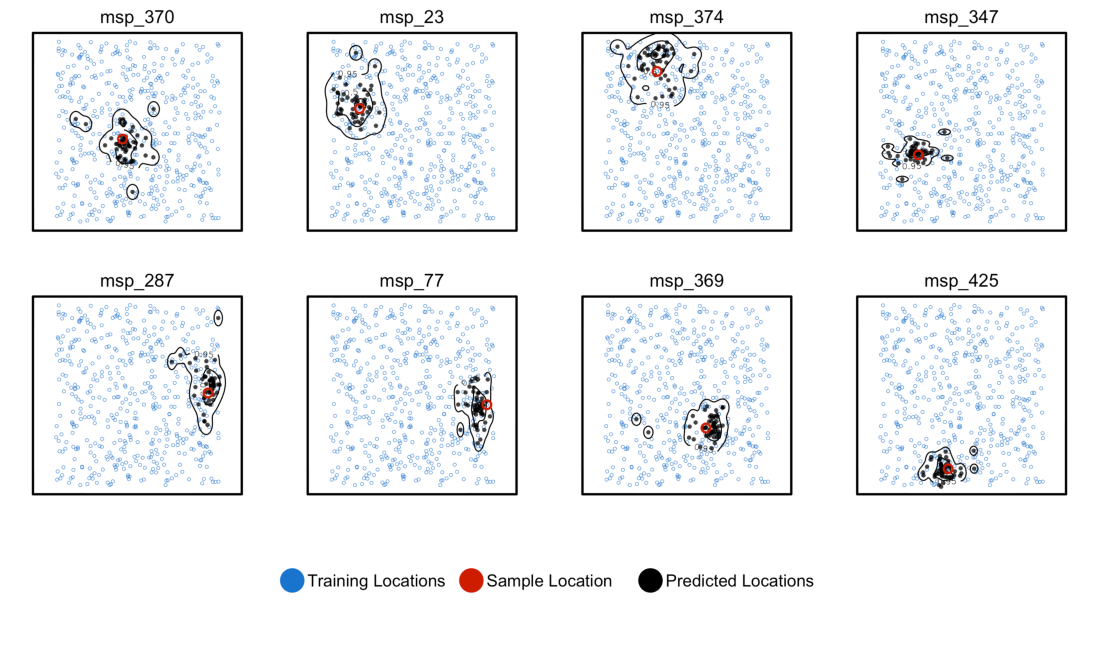
from Battey et al 2020
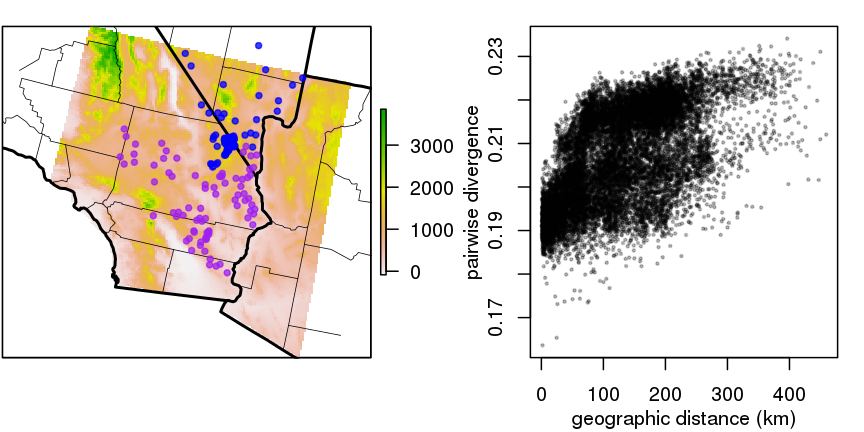
- genetic versus geographic distance between pairs of 272 desert tortoises (McCartney-Melstad, Shaffer)
- clouds are comparisons within/between the two colors
Software development goals
- open
- welcoming and supportive
- reproducible and well-tested
- backwards compatible
- well-documented
- capacity building
Thanks!
- Andy Kern
- Matt Lukac
- Murillo Rodrigues
- Victoria Caudill
- Anastasia Teterina
- Jeff Adrion
- CJ Battey
- Jared Galloway
- the rest of the Co-Lab
Funding:
- NIH NIGMS
- NSF DBI
- Sloan foundation
- UO Data Science
- Jerome Kelleher
- Ben Haller
- Ben Jeffery
- Georgia Tsambos
- Jaime Ashander
- Gideon Bradburd
- Madeline Chase
- Bill Cresko
- Alison Etheridge
- Evan McCartney-Melstad
- Brad Shaffer
- Sean Stankowski
- Matt Streisfeld
![]()
![]()